
سندرم آلپورت
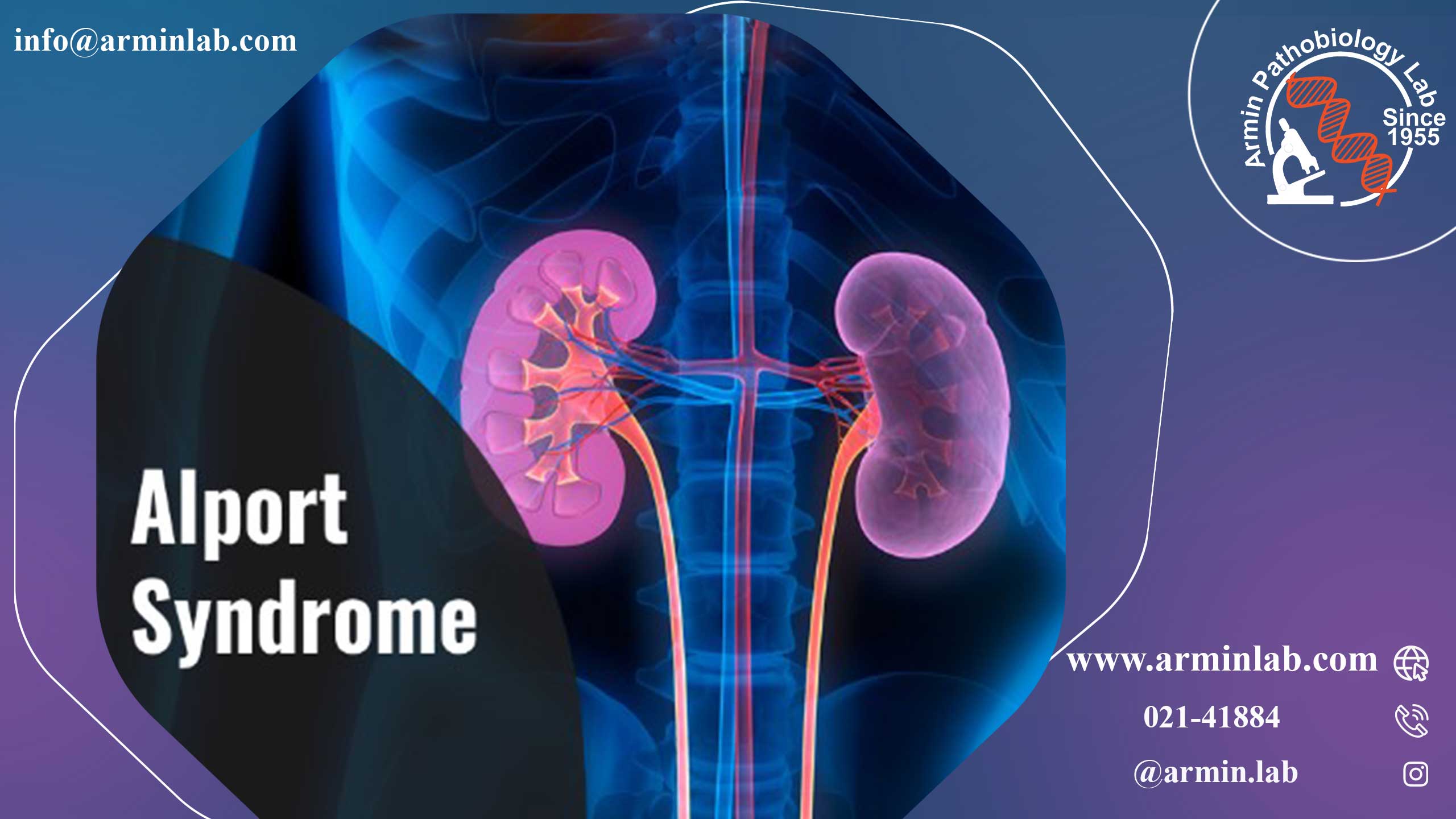
سندرم آلپورت یک بیماری ژنتیکی است که در آن کلیه های فرد پروتئین های کلاژن نوع IV طبیعی را تولید نمی کنند.
کلاژن نوع IV از سه زنجیره کلاژن مجزا (زنجیره آلفا) تشکیل شده است که مانند طناب به هم می پیچند. این زنجیرها آلفا 3، آلفا 4 و آلفا 5 هستند. اگر بدن یکی از این زنجیره ها را تولید نکند، دو زنجیره دیگر نمی توانند ترکیب شوند و این موضوع منجر به ایجاد شدیدترین علائم سندرم آلپورت می گردد.
اگر بدن همه ی زنجیرهها را تولید کند اما یکی را به درستی نسازد، گاهی اوقات زنجیرهها نمیتوانند با هم ترکیب شوند و گاهی ترکیب میشوند اما به درستی عمل نمیکنند. علائم سندرم آلپورت در این موارد ممکن است خفیف تر باشد.
کلاژن نوع IV یک پروتئین مهم در غشاهای تصفیه در کلیه ها (غشاء پایه گلومرولی یا GBM) است.
GBM بخشی از یک ساختار سه لایه است که خون فرد را تصفیه می کند تا سموم و سایر محتویاتی را که بدن برای تولید ادرار به آن نیاز ندارد از بین ببرد. GBM همچنین از باقی ماندن محتویاتی مانند سلول های خونی و پروتئین ها درخون به جای ورود به ادرار اطمینان حاصل می نماید.
درصورتی که GBM به درستی فعالیت نداشته باشد، خون یا پروتئین می تواند به ادرار نشت کند. توانایی کلیهها برای فیلتر کردن ادرار نیز با گذشت زمان بدتر میشود که خطر نارسایی کامل کلیه را افزایش میدهد.
کلاژن نوع IV نیز در گوش و چشم فرد وجود دارد. بنابراین اگر به سندرم آلپورت مبتلا هستید، ممکن است در بینایی و شنوایی و همچنین کلیه ها مشکل داشته باشید.
سندرم آلپورت سه نوع ژنتیکی دارد، که عبارتند از:
-
سندرم آلپورت مرتبط با کروموزوم (X)(XLAS) :
XLAS با کروموزوم X است. کروموزوم X یکی از دو کروموزوم جنسی فرد (X و Y) و محلی است که ژن تولید کننده زنجیره آلفا 5 (COL4A5) در آن قرار دارد.
مردان دارای یک کروموزوم X و یک کروموزوم Y هستند. زن ها دو کروموزوم X دارند. مردان مبتلا فقط کروموزوم X غیرطبیعی دارند، بنابراین به احتمال زیاد علائم شدیدتری را تجربه می کنند. زنان مبتلا دارای یک کروموزوم X غیر طبیعی و یک کروموزوم X طبیعی هستند، بنابراین علائم آنها معمولا خفیف تر است.
مردان کروموزوم Y خود را به فرزندان پسر خود منتقل می کنند. بنابراین، آنها نمی توانند XLAS را به پسران منتقل کنند. با این حال، مردان کروموزوم X خود را به همه فرزندان دختر خود منتقل می کنند. در نتیجه همهی فرزندان دختر آنها به سندرم آلپورت مبتلا خواهند شد.
زنان یکی از کروموزوم های X خود را به فرزند خود از هر جنس منتقل می کنند. بنابراین، آنها 50 درصد شانس انتقال XLAS به هر یک از فرزندان خود را دارند.
XLAS شایع ترین نوع سندرم آلپورت است و حدود 60 تا 80 درصد از کل موارد سندرم آلپورت را تشکیل می دهد.
-
سندرم آلپورت اتوزومال مغلوب (ARAS):
اصطلاح “اتوزومال” به بیماری اشاره دارد که برای ایجاد شدن به جهش در هر دو ژن به صورت جفتی نیاز دارد. “اتوزومی” به 23 جفت ژن اتوزومال اشاره دارد. اتوزومال مغلوب یک الگوی توارثی است. اگر والدین دارای صفت اتوزومال مغلوب باشند، هیچ علامتی نشان نمی دهند. دراین حالت، برای اینکه والدین این بیماری را به فرزندان خود منتقل کنند، می بایست هر دو مبتلا باشند. با این حال، از آنجایی که هیچ علامتی ندارند، اغلب حتی نمی دانند که به آن مبتلا هستند. هر دو والدین باید یک ژن تغییر یافته را به فرزندشان منتقل کنند تا کودک شرایط یا صفت ژنتیکی را در الگوی اتوزوم مغلوب به ارث ببرد. اگر هر دو والد دارای ویژگی اتوزوم مغلوب باشند، فرزاندان آنها به احتمال 25 درصد مبتلا می شوند. فقط تغییراتی که در DNA اسپرم یا تخمک رخ می دهد می تواند از والدین به فرزندان منتقل شود.
در سندرم آلپورت، ژن هایی که پروتئین های آلفا 3 (COL4A3) و آلفا 4 (COL4A4) را کد می کنند در کروموزوم 2 قرار دارند.
در ARAS، یک جهش در هر دو ژن در کروموزوم 2 وجود دارد که پروتئین های آلفا 3 یا آلفا 4 را کد می کند. ARAS به جنسیت فرد بستگی ندارد، بنابراین وراثت و شدت ARAS در همه ی افراد یکسان است.
اگر مبتلا به ARAS هستید، احتمال انتقال یکی از ژن های غیر طبیعی به هر یک از فرزندان بیولوژیکی 50 درصد است. این موضوع معمولاً منجر به ایجاد سندرم آلپورت نمی شود. 25 درصد احتمال انتقال هر دو ژن غیر طبیعی به فرزندان بیولوژیکی وجود دارد که در این صورت، فرزند به ARAS مبتلا خواهد بود.
ARAS حدود 15 درصد از کل موارد سندرم آلپورت را تشکیل می دهد.
-
سندرم آلپورت اتوزومال غالب (ADAS):
“غالب” به نوعی بیماری اشاره دارد که برای ایجاد آن، فقط به یک جهش در یکی از ژن ها در یک جفت، نیاز است. در ADAS، یک جهش در یکی از ژنهای کروموزوم 2 وجود دارد که پروتئینهای COL4A3 یا COL4A4 را کد میکند.
ADAS به جنسیت بستگی ندارد، بنابراین وراثت و شدت آن در همه ی افراد یکسان است.
درصورت ابتلا به این نوع، 50 درصد احتمال انتقال ژن غیر طبیعی به فرزندان بیولوژیکی وجود دارد که در این صورت ADAS ایجاد می شود.
ADAS 25 تا 35 درصد از کل موارد سندرم آلپورت را تشکیل می دهد.
 سندرم آلپورت چه کسانی را تحت تاثیر قرار می دهد؟
سندرم آلپورت چه کسانی را تحت تاثیر قرار می دهد؟
سندرم آلپورت می تواند هر کسی را تحت تاثیر قرار دهد.
این بیماری از نوع ارثی است، به این معنی که یک یا هر دو والدین بیولوژیکی آن را به فرزندشان منتقل می کنند. با این حال، در حدود 15٪ موارد، زمانی که هیچ یک از والدین ژن جهش یافته را نداشته باشند، می تواند ایجاد شود.
شیوع:
پزشکان معتقد هستند که سندرم آلپورت نادر است.
محققان شیوع آن را 1 در هر 50000 تولد زنده در سرتاسر جهان تخمین می زنند. با این حال، با مطالعه ی بیشتر محققان در این زمینه ، افراد بیشتری با اشکال خفیف تر این بیماری شناسایی می شوند. در نتیجه، سندرم آلپورت ممکن است شایعتر از آن چیزی باشد که دادههای موجود در حال حاضر نشان میدهند.
علائم و علل
سندرم آلپورت چگونه باعث نارسایی کلیه می شود؟
درصورت ابتلا به این بیماری، غشاهای پایه گلومرولی به درستی فیلتر نمی شوند، بنابراین خون و پروتئین به ادرار نشت می کند. با این حال، از سلول هایی که هر دو طرف آن را پوشاندهاند نیز پشتیبانی نمی کند. در نتیجه سلول ها تحریک شده و ملتهب می شوند. پودوسیت ها سلول هایی هستند که GBM ها را تولید می کنند و زمانی که سلول های اطراف ملتهب می شوند، سعی می کنند کلاژن نوع IV بیشتری را در GBM ها قرار دهند. وقتی این اتفاق می افتد، GBM ها ضخیم تر و بی نظم می شوند. این موضوع باعث نشت پروتئین به ادرار یا حالت پروتئینوری می گردد.
با گذشت زمان و با نشت بیشتر پروتئین به ادرار و ضخیم تر شدن GBM، ممکن است بافت اسکار یا فیبروز ایجاد شود. در نتیجه، توانایی کلیه ها برای تصفیه خون شروع به کاهش می کند (بیماری مزمن کلیه). با ایجاد بافت اسکار بیشتر، عملکرد کلیه وخیم تر می گردد تا زمانی که در نهایت کلیه ها از کار بیفتند ( حالت نارسایی کلیه).
علائم بالینی اصلی
- علائم سندرم آلپورت ممکن است بسته به نوع آن در افراد مختلف متفاوت باشد. اما علائم اصلی آن عبارتند از:
- خون در ادرار که قابل مشاهده نیست (هماچوری میکروسکوپی)
- پروتئین در ادرار (پروتئینوری)
- بیماری مزمن یا نارسایی کلیه
- کم شنوایی
- مشکلات چشم
اولین علامت سندرم آلپورت، هماچوری میکروسکوپی است. هماچوری میکروسکوپی به این دلیل رخ می دهد که GBM های غیر طبیعی گلبول های قرمز خون را به ادرار نشت می دهند. سلول های خونی با چشم غیرمسلح قابل دیدن نیستند و فقط با استفاده از میکروسکوپ قابل مشاهده اند. مردانی که به XLAS مبتلا هستند و هرکسی که به ARAS مبتلا است در بدو تولد دچار هماچوری میکروسکوپی خواهد شد. اکثر زنانی که XLAS دارند به مرور زمان دچار هماچوری میکروسکوپی می شوند. همه افراد مبتلا به ADAS دچار هماچوری میکروسکوپی نمی شوند.
هنگامی که عملکرد کلیه شروع به کاهش می کند، بیماری مزمن کلیه (CKD) ایجاد می شود. بیشتر افراد تا زمانی که به نارسایی کلیه نرسند، علائم بیماری مزمن کلیه را ندارند.
علائم نارسایی کلیه عبارتند از:
- تورم (ادم)، به خصوص در اطراف دست یا مچ پا
- خستگی شدید
- حالت تهوع و استفراغ
- گرفتگی عضلات
کم شنوایی در مردان مبتلا به XLAS و افراد مبتلا به ARAS شایع تر است. با این حال، ممکن است در افراد مبتلا به سندرم آلپورت رخ دهد. کاهش شنوایی معمولاً تدریجی است. بسیاری از مردم تا زمانی که شدت بیشتری پیدا کند متوجه نمی شوند که دچار کم شنوایی هستند. اکثر مردم در شنیدن صداهای بلندتر دچار مشکل هستند، هرچند ممکن است برخی افراد در نهایت قادر به شنیدن هیچ صدایی نباشند. ممکن است در نهایت به استفاده از سمعک نیاز باشد. در موارد شدید، کم شنوایی کامل (ناشنوایی) رخ می دهد.
ممکن است طیف وسیعی از مشکلات در چشمان فرد ایجاد گردند. سطح چشم در برخی از افراد بیشتر احتمال دارد که دچار خراشیدگی شود (ساییدگی قرنیه) که ممکن است زمان بهبودی آن مدت زیادی طول بکشد. ساییدگی قرنیه ممکن است باعث آبریزش چشم و درد شود، اما معمولاً منجر به از دست دادن بینایی در فرد نمی گردد. برخی از افراد همچنین در قسمت شفاف چشم خود که به تمرکز بینایی آنها کمک می کند (عدسی) دچار مشکل می شوند که در نهایت می تواند ایجاد آب مروارید کند.
آیا سندرم آلپورت مسری است؟
خیر، سندرم آلپورت مسری نیست و نمی تواند از طریق تماس نزدیک به فرد دیگری منتقل شود.
سندرم آلپورت یک بیماری ارثی است، به این معنی که یکی یا هر دوی والدین بیولوژیکی باید آن را به فرزند منتقل کنند.
تشخیص و آزمایشات:
اگر به هماچوری میکروسکوپی یا بیماری مزمن مبتلا هستید، پزشک متخصص ممکن است سندرم آلپورت را به عنوان یک مورد نگرانی مطرح نماید. اگر سابقه خانوادگی بیولوژیکی سندرم آلپورت دارید، غربالگری می تواند به تشخیص آن کمک کند. درصورت عدم وجود سابقه ی خانوادگی، پزشک می تواند بر اساس سابقه و آزمایشات اضافی بیماری را تشخیص دهد.
پزشک علائم فرد را بررسی می کند و در مورد سابقه خانوادگی بیولوژیکی وی می پرسد. انواع آزمایشها نیز میتوانند به پزشک در تشخیص سندرم آلپورت کمک کنند.
این تست ها عبارتند از:
آزمایش ادرار: آزمایش ادرارجنبه های ظاهری، شیمیایی و میکروسکوپی ادرار را بررسی می کند. این آزمایش می تواند به پزشک کمک کند تا خون یا پروتئین موجود در ادرار فرد را شناسایی کند.
آزمایش کلیرانس کراتینین یا آزمایش خون سیستاتین C: این آزمایشها سطح کراتینین و پروتئین سیستاتین C در خون را اندازهگیری میکنند. آزمایشها نشان میدهند که کلیهها چقدر دارای توانایی تصفیه ی خون هستند.
نرخ فیلتراسیون گلومرولی تخمینی (:(eGFR مقداری است که از کراتینین یا سیستاتین C محاسبه می شود و تخمین میزند که کلیههای فرد چقدر قابلیت تصفیه خون را دارا هستند.
بیوپسی کلیه: چندین تکه کوچک از بافت کلیه شما برای بررسی زیر میکروسکوپ در آزمایشگاه برداشته می شد. نمونه ها می توانند الگوهای مختلفی را نشان دهند که بر کلیه شما تأثیر می گذارد. در سندرم آلپورت، GBM های غیر طبیعی نازک به نظر می رسند، اما ممکن است شامل مناطق ضخیم شوند. درصورتی که سندرم آلپورت شدیدتر باشد، واحدهای فیلتر و ساختار پشتیبانی ممکن است اسکار یا جای زخم را نشان دهند.
آزمایش ژنتیک: یک آزمایش ژنتیکی می تواند جهش در ژن های کلاژن را شناسایی کند.
تست شنوایی: پزشک ممکن است در صورت مشکوک شدن به سندرم آلپورت، آزمایش شنوایی(ادیوگرافی) را درخواست دهد. فرد می بایست هر چند سال یک بار آزمایشاتی را که نشان دهنده ی ایجاد کم شنوایی یا وخامت آن است انجام دهد.
معاینه چشم: متخصص چشم معاینه چشم را انجام می دهد و سطح چشم (قرنیه)، عدسی و پشت چشم (شبکیه) مشاهده می کند تا بر تاثیر سندرم آلپورت بر این نواحی آگاه شود. همچنین ممکن است یک آزمایش تصویربرداری توموگرافی انسجام نوری (OCT) انجام دهند.

مدیریت و درمان
آیا سندرم آلپورت قابل درمان است؟
هیچ درمانی برای سندرم آلپورت وجود ندارد. محققان در حال مطالعه بر روی روش های ژن درمانی هستند که ژن های غیرطبیعی را برطرف می کنند، اما هنوز موفق نشده اند. درصورت موفقیت در ایجاد یک روش ژن درمانی، این روش برای سال های زیادی در دسترس نخواهد بود. با این حال، اکنون درمان هایی در دسترس هستند که زوال کلیه را کاهش داده و نارسایی آن را به تاخیر می اندازند.
چگونه می توانم از سندرم آلپورت جلوگیری کنم؟
سندرم آلپورت قابل پیشگیری نیست، اما آگاهی از سابقه خانوادگی می تواند به تشخیص زودهنگام کمک و همچنین از انتقال آن به فرزند بیولوژیکی فرد جلوگیری کند.
تشخیص زودهنگام سندرم آلپورت و شروع درمان بهترین راه برای به تاخیر انداختن نارسایی کلیه است. اگر پزشک خون را در ادرار تشخیص دهد، بهتر است آزمایشات بیشتری برای سندرم آلپورت انجام شود، به خصوص اگر مشکل شنوایی یا کاهش عملکرد کلیه وجود داشته باشد.
درصورت وجود سابقه ی خانوادگی هماچوری، پزشک باید ادرار را از نظر خون آزمایش کند و آزمایش خون را برای بررسی عملکرد کلیه تجویز نماید.
مراقبت از خود:
در صورت ابتلا به سندرم آلپورت پزشک کمک می کند تا بهترین برنامه درمانی را برای فرد ایجاد کند، که ممکن است شامل داروها یا تغییر در شیوه زندگی شما باشد.
زمان مناسب مراجعه به پزشک:
در صورت وجود خون در ادرار، کاهش شنوایی یا کاهش بینایی با یک فرد متخصص مشورت کنید. این موارد می توانند نشانه های سندرم آلپورت باشند.
در صورت ابتلا به این بیماری، مطمئن شوید که پزشک شما را به یک متخصص کلیه (نفرولوژیست) ارجاع دهد.
درصورت وجود سابقه ی این بیماری در عضوی از خانواده، می توانید با پزشک مشورت نمایید.
مطالب مشابه:
عفونت کلیه
سندرم روده کوتاه
عفونتهای نفاس
بارداری و بیماری کلیوی
تستهای عملکرد کلیه (KFT)
منبع: my.clevelandclinic






1 دیدگاه